
This website uses cookies to improve your experience. We'll assume you're ok with this, but you can opt-out if you wish. Privacy & Cookies policy








|

|
|
|---|---|---|
|
Select if |
You are working with commercial or proprietary libraries of millions of compounds |
If you want to explore libraries such us Enamine REAL and obtain synthesizable compounds |
|
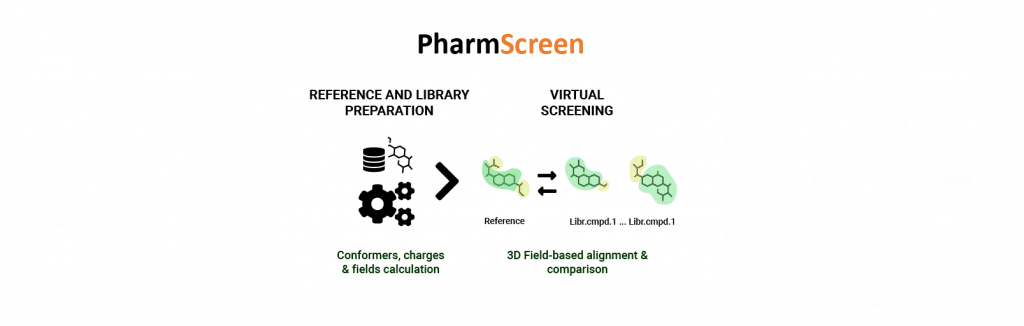
Descriptors |
3D QM-derived hydrophobic molecular field descriptors |
|
|
Library Size |
Millions of Compounds |
Billions of Compounds |
|
Search Engine |
Full Library Enumeration |
Building Blocks |
|
Delivered Molecules |
Synthesizable compounds from vendors or from your libraries |
Synthesizable compounds from building blocks
|
|
|
|
|---|---|
|
Choose for commercial and proprietary libraries screening up to 10M |
Choose to explore libraries such us Enamine REAL and obtain synthesizable compounds |
|
QM-derived hydrophobic molecular field descriptors |
|
|
Few Millions of Compounds |
Billions of Compounds |
|
Full Library Enumeration Search Engine |
Building Blocks Search Engine |
|
Purchasable compounds from vendors |
Synthesizable compounds
|